

FDA于2017年9月28日发布了关于抗肿瘤药物生殖毒性试验和说明书要求的工业化技术指南征求意见稿,请社会公众在60天内提出修订建议。我国目前也有大量抗肿瘤新药在注册申请,也面临着同样的技术问题。为此,药理毒理学部深入学习了该指南内容,并整理了该指南的中文翻译稿。后续我们将进一步跟踪国外相关技术要求进展,汇总国内抗肿瘤药物上市申请中的生殖毒性支持数据信息,并征求国内行业的意见和建议,明确我国抗肿瘤药物新药上市申请的生殖毒性风险评价要求。
1. 简介
本指导原则的目的是帮助申请人进行抗肿瘤药物生殖毒性评估[主要是胚胎-胎仔发育毒性(EFD)],并推荐在说明书中标注停止治疗后的避孕时间,使对胚胎/胎儿发育的潜在风险最小化。本指导原则将在以下方面进行讨论。
● 各类药物的EFD毒性评价,以及何时不需要进行这些试验。
● 针对特定人群药物的EFD毒性评价。
● 使用非临床信息(如遗传毒性和一般毒性试验的结果),评估进行专门的EFD试验的的必要性。
● 说明书中关于EFD试验和人体发育不良结果潜在风险(说明书中妊娠部分)的建议,以及关于患者避孕以最小化胚胎/胎儿(说明书中男女生殖潜力部分)发育风险的建议。
本指导原则的药物是指小分子、治疗性蛋白、抗体和相关制品如偶联产品。本指导原则不阐述生物类似药、可互换药物、放射性药物、细胞和基因治疗产品、癌症疫苗的风险。术语“致畸性”是指对正常胚胎-胎仔发育的干扰从而可能导致畸形和死亡的事件。然而,某些类型药物(例如肿瘤免疫)的胚胎-胎仔致死性可能不是由于药物对胎仔的直接作用所致,而是由于免疫排斥,并不具有明显的致畸性。因此,本指导原则中的胚胎-胎仔致死性包括所有原因引起的死亡,不管是否存在畸形。
本指导原则不涉及暴露量或剂量的安全范围。很多抗肿瘤药物特别是本指导原则所涉及的小分子药物,未确定安全剂量范围(即在与人体推荐剂量相当或低于人体推荐剂量的暴露时,在动物中观察到胚胎-胎仔毒性)。胚胎-胎儿的发育风险是在患者中主要担忧的问题,也是进行EFD试验的原因,以便可能在药品说明书中包括关于避孕的合适建议。然而,本指导原则不涉及临床试验中胚胎-胎儿的潜在风险,因为相关的药物开发过程需要采取足够的避孕措施。虽然对于晚期肿瘤适应症的上市申请一般不需要生育力试验和围产期试验,本指导原则包含了在非晚期肿瘤适应症中讨论了以上两方面的一些内容。
本指导原则是ICH S9(抗肿瘤药物的非临床评价)的补充。ICH S5(药物生殖毒性和雄性生育力毒性检测)和ICH S6(R1)(生物制品的临床前评价)阐述了生殖毒性评价的特殊试验设计。本指导原则提供了一些目前S9没有描述的和S6仅简单讨论的替代性评价的案例。本指导原则还提供了也提供了目前S9未涉及的关于药物潜在生殖毒性风险和避孕的非临床建议。
一般来说,FDA指导原则文件不具备强制的法律效应。指导原则代表目前FDA对于该主题的考虑,应该被视作仅为建议,除非注明为特别的监管或法规要求。监管机构的指导原则中的“应(should)”指的是建议或推荐,而不是要求。
2 .背景
ICH S9推荐了抗肿瘤药物IND申请和后续开发的非临床研究所需类型和时间安排。对于S9范围类的药物,该指导原则建议,在新药申请上市(NDA)或新生物制品申请上市时提交EFD试验结果。
有些情况可能不需要EFD毒性试验。例如,具有遗传毒性且一般毒性试验确定靶向于迅速分化细胞的药物(ICH S9),一般认为可引起致畸性或胚胎-胎仔死亡。在另外一些情况下,作为EFD试验的替代,可提供替代性的风险评估。自ICH S9发布以来,FDA已经获得了抗肿瘤药物替代动物生殖毒性试验进行生殖毒性评估的替代方法的经验。
目前ICH或FDA指导原则对抗肿瘤药物没有包括避孕的建议。由于抗肿瘤药物的毒性特点,有必要采用避孕措施来最小化发育中胚胎暴露风险。
3. 胚胎-胎仔发育毒性评价
3.1 一般建议
一般情况下,当重点担忧胚胎-胎仔发育毒性时,生殖毒性试验应遵循ICH S9的建议。EFD试验应该在两个动物种属中进行,一般为大鼠(或小鼠)和兔,除非在一个动物种属中具有致畸性或胚胎-胎仔死亡为阳性,这时可能不需要进行第二个种属试验。在一些情况下,非GLP初步试验明确地表明存在胚胎-胎仔死亡或致畸风险时,可能不需要确证性的GLP试验。
3.2 细胞毒类药物
具有遗传毒性且一般毒性试验确定靶向于迅速分化细胞的药物,一般认为具有致畸性或胚胎-胎仔致死性。这种情况下,EFD试验被认为是不必要的。在确定EFD试验必要性时,必须至少两种遗传毒性试验的结果为阳性才能作为该药物具有遗传毒性的判断。
3.3 生物制品
参照ICH S9,应在一种药理学相关动物种属中进行EFD试验。当药理学相关种属是非人灵长类时,应考虑ICH S6(R1)所描述的增强性围产期毒性试验(ePPND),试验设计参考ICH S6(R1)。当对于该临床候选物没有药理学相关动物种属时,可考虑使用良好表征和生物相关的替代产品(如果有)。然而,一般不必仅为了EFD试验而单独制备替代产品。
当EFD试验不必要时,应该完成替代评估。评估应包括以下信息或数据:
● 文献评估。评估应包括以下资料和信息:
— 描述胚胎-胎仔中的靶点表达。
— 描述分子靶点在胚胎-胎仔发育中的作用。
— 如果可获得,包括来源于基因敲除、转基因动物或突变基因动物的数据(当合适时)。
— 基于前面列出的目标项,描述受试物的影响,如妊娠的丢失或子代中的表型特征。
● 体外研究如透过胎盘屏障的能力(如果未知)、与胚胎-胎仔组织的交叉反应。评估应描述由于靶结合而引起的对发育的潜在影响。
(虽然本部分内容是对生物制品而言,但合适时也适用于小分子药物。)
3.4 偶联药物
对于包含有生物制品部分和小分子部分的偶联药物,EFD试验的设计取决于多种因素,如生物制品部分对靶点的结合、小分子释放的可能、小分子的性质(例如作用机制和细胞毒性)、对毒性来源的认知(来源于生物制品部分还是小分子部分)。例如抗体偶联(ADC)药物,当小分子部分是细胞毒类药物(遗传毒性和靶向于迅速分化细胞)时,不需要进行EFD试验(见第3.2项目部分)。当ADC药物需要进行EFD试验时,如果ADC药物的毒性来自于小分子,而抗体部分不与动物种属中的靶点结合,那么该EFD试验可以用小分子进行。当偶联药物生物制品部分与动物种属中的靶点结合时,通常推荐用偶联药物进行生殖毒性试验。
3.5 联合用药
当两种药物需要联合应用(如21CFR 3.2)时(该两种药物对于达到拟定应用、适应症或疗效均必需),EFD试验时也联合应用。如果一种药物已经有EFD数据,且显示具有致畸性或胚胎-胎仔致死性时,不必要再进行额外的联合应用的EFD试验。
3.6 脂质体产品
通常脂质体制剂改变了活性药物成分(API)的药代动力学参数(如增加暴露量)。如果未包封药物以前进行了EFD试验,且可见致畸性和/或胚胎-胎仔致死性,那么可能不必再进行单独的脂质体制剂EFD试验。但是,当API未见致畸性或胚胎-胎仔致死性时,需要进行脂质体制剂EFD试验,因为脂质体制剂增加的暴露量和脂质体制剂中新的成分可能会影响胚胎-胎仔发育。申请人考虑第3项目下适用情况时取决于脂质体包封的性质。例如,当脂质体包封细胞毒类药物时,申请人应考虑参照第3.2项目的细胞毒类部分的要求。
4 生育力评价
ICH S9中的治疗晚期癌症患者的药物一般不必进行单独的生育力和早期胚胎毒性研究。一般毒性试验中对雄性和雌性生殖器官的评价,以及其它相关终点(例如性激素的改变),在评估药物对生育力潜在作用时应予考虑。从这些观察中所确定的生育力风险应描述在说明书的“致癌性、致突变性、生育力损伤”部分,并总结在说明书的“男性和女性生殖潜在影响“的部分。
当适应症不是晚期肿瘤时,一般需要进行单独的生育力试验。基于总体数据分析,如果单独的生育力试验不会提供有用的信息,那么不必要进行单独的生育力试验。例如,用于早期前列腺癌患者的药物,抑制雄性激素达到去势水平,不需要进行雄性动物(因为药物被认为会导致不育)或雌性动物(因为是雄性专用药)的生育力试验。此外,如果一般毒性试验发现提示了对生育力的不良影响(例如精子数减少或者卵泡丢失),通常不需要再进行单独的生育力试验。
临床试验中睾丸毒性的评价(如FDA“药物开发过程中的睾丸毒性评价指导原则草案”所提到的)不是必需的。因为由于抗肿瘤药物的毒性,其临床试验一般不在健康受试者中进行,所推荐的试验试验在肿瘤患者中特别不可行。
5 围产期毒性评价
ICH S9中的治疗晚期癌症患者的药物可能不需进行围产期毒性试验。然而,当围产期试验被认为是必要时(基于适应症),应考虑这种试验是否能为患者或医师提供信息。如以下案例:
● 致畸性药物可能不需进行围产期毒性试验。这种药物预期会对生存和一般健康产生不利影响,包括后代的生长和发育,且这种风险应该在说明书的“妊娠”部分进行描述。
● 对于引起胚胎-胎仔死亡的药物,应考虑是否有足够数量的子代来评价对发育的影响。对于引起胚胎-胎仔死亡的药物,可能需考虑改良的围产期试验以增加活胎数,比如短时间窗内给药。试验计划的修改不应改变围产期试验的目的(例如,在出生后开始给药将只能提供产后生长的信息,因此这样不是必需的)。
6 特定人群风险评价
6.1 仅用于男性的药物
由于EFD试验是研究胚胎-胎仔发育毒性的风险,所以对于男性专用药物(如前列腺癌),不需要进行EFD试验。如第3.1项目的一般建议部分所讨论的,评价精液转移对于发育中的胚胎的风险不是必需的,而是推荐一段时间的避孕(见第8项目的避孕推荐)。避孕信息需要在说明书的“男性和女性生殖潜在影响”部分予以标明。对于这种患者不必进行围产期毒性试验。对于非晚期肿瘤适应症(例如早期前列腺癌)应考虑进行雄性动物生育力试验(见第4项目的生育力评价部分)。
6.2 仅用于绝经后妇女的药物
仅用于绝经后妇女的抗肿瘤药物不必进行生殖毒性试验。通常, “绝经”定义为无替代医学原因长期停经超过12个月,或根据其它因素定义,如血清促卵泡激素(FSH)水平和双侧卵巢切除术。然而,这个定义及其对于拟定临床试验项目的适用性应该与FDA相关的临床审评部门讨论。
6.3 儿科用药
S9中的晚期癌症用药,当适应症包括达到青春期患者时,需要进行EFD试验或EFD评价(如果合适)。这通常包括女性和男性的生殖潜力的评估,且包括青少年(12~18岁)。当用药旨在治愈或大幅度提高生存期时,应考虑进行完整的生殖毒性组合试验(即生育力、EFD、围产期毒性试验),除非该药属于前面已经讨论过的可能不需要进行生殖毒性试验的情况(见第3项目到第4.1项目部分)。
7 药代动力学参数
7.1 不成比例的代谢产物
对于人体特异性代谢产物,或者与动物毒性试验比较人体中不成比例高水平代谢产物,可能需要额外的代谢产物EFD试验。应考虑在动物EFD试验中的动物种属中是否具有充分的暴露,代谢产物的结果包含在API试验结果中。当API具有胚胎-胎仔致死性或致畸性时,代谢产物不需要进行EFD试验。
7.2 暴露量比较
EFD试验应收集药代动力学数据,且说明书的“妊娠”部分应标明动物与人的曲线下面积(AUC)比例。当EFD试验未获得药代动力学参数时,当合适时(例如,基于制剂的差异)可以采用来自于采用相同的动物种属、剂量、给药途径、给药方案的一般毒性试验中的动物AUC 。
8 避孕推荐
当确定抗肿瘤药物的发育毒性风险之后,说明书中应向患者提供停止治疗后的避孕时间信息。说明书的“女性和男性生殖潜在影响”部分需要包含推荐的男性和女性用药后的避孕期,以最小化EFD风险和接受抗肿瘤药物治疗男性患者的女性伴侣的风险。
下述8.1遗传毒性药物和8.2非遗传毒性药物部分中的建议的科学基础,是基于对配子发生和此过程中的性别差异的认识。这些推荐基于发育毒性(比如畸形和致死性)的预防,而不是生育力的恢复。
虽然以下推荐旨在减少母体药物的暴露,但也可以通过减少代谢产物的暴露而降低发育毒性(例如遗传毒性代谢产物)。
8.1 遗传毒性药物
8.1.1 男性
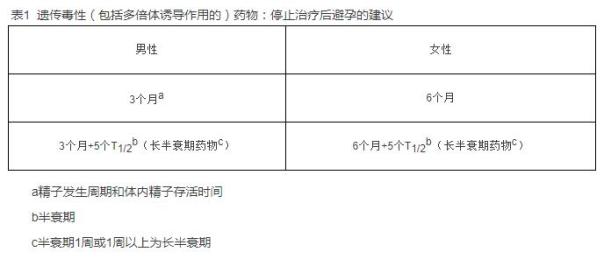
遗传毒性药物可能造成精子中的脱氧核糖核酸(DNA)损伤,从而有可能对其女性伴侣的胚胎产生不良影响。虽然还没有关于使用抗肿瘤药物男性患者后代畸形率上升的报道,但是可以在给予抗肿瘤药物的雄性动物和正常雌性动物交配的试验中观察到这种作用。抗肿瘤治疗停止后3个月的避孕期将最小化半衰期较短(少于1周)的遗传毒性药物对胚胎-胎仔的不良影响。人体精子发生周期大约为70天。3个月考虑了药物的半衰期和体内精子的存活时间。对于较长半衰期(1周或1周以上)的药物,推荐再增加5个半衰期的避孕期。见表1。
8.1.2女性
遗传毒性药物可能直接影响胚胎/胎儿,或可能造成卵母细胞的DNA损伤。卵泡生长周期大约是6至12月。在起始阶段(原始卵泡)暴露于遗传毒性药物将主要导致卵泡丢失。保留在卵泡中的损伤可能通过自然的卵泡闭锁过程而被进一步清除(清除率为90%以上)。卵泡生长的生长期和成熟期(4~6个月)对持续的DNA损伤是最敏感的,很可能导致胚胎-胎仔畸形。因此,遗传毒性药物停止治疗后推荐6个月的避孕期。对于较长半衰期(1周或1周以上)的药物,推荐增加5个半衰期的避孕期。见表1。
8.2 非遗传毒性药物
8.2.1 男性
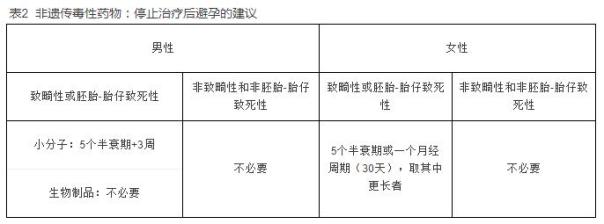
精液中药物的存在具有理论上的致畸风险。虽然有报道提示使用抗肿瘤药物的男性的后代畸形率没有上升,但是没有专门考察停止治疗第一年的出生情况的报道。2014年发表的科技文章表明包括沙利度胺在内的药物阴道内给药,在临床相关浓度下未造成胚胎畸形。尽管如此,更早期的研究观察到雄性兔给予沙利度胺对胚胎-胎仔产生不良影响。虽然沙利度胺不会在精液中蓄积,但是许多小分子药物可以在精液中蓄积,而且关于精液转移造成的胚胎-胎仔毒性的研究还比较有限。由于存在数据缺口,对于小分子致畸性药物,推荐5个半衰期再加上3周(考虑到体内精子的药物存留)作为避孕期。但是,对于致畸性生物制品,对于男性不推荐避孕期,因为这类生物制品不会在精液中蓄积、吸收有限、可能因阴道和子宫颈的酶而蛋白降解。见表2。
8.2.2女性
停止治疗后避孕5个半衰期可以使大约97%的发育毒性药物在受精前从循环系统中清除。对于短半衰期的药物,推荐停止治疗后至少30天(一个月经周期)的避孕期。见表2。
翻译: 叶旋
校对:黄芳华、王海学
参考文献:Oncology Pharmaceuticals:Reproductive Toxicity Testing and Labeling Recommendations Guidance for Industry。FDA guideline,09/28/2017
来自:CDE,国际药事通道 国际药事法规更新
原标题:FDA关于抗肿瘤药物生殖毒性试验和说明书建议的指导原则(草案)